An ecology research led by Prof. Douglas YU and YANG Chunyan from Kunming Institute of Zoology of the Chinese Academy of Sciences present a co-designed wet-lab and bioinformatic workflow, Biodiversity soup II, for metabarcoding bulk samples that removes both false-positive (tag jumps, chimeras, erroneous sequences) and false-negative (‘dropout’) errors. The paper was published in Methods in Ecology and Evolution (MEE) on 30th March,2021.
In 2012, Douglas YU’s group published a metabarcoding paper entitled “Biodiversity soup: metabarcoding of arthropods for rapid biodiversity assessment and biomonitoring” in MEE. This paper was one of the first to describe a metabarcoding protocol, and it became one of the most downloaded papers in this journal.
Since then, there has been a flood of basic research in metabarcoding methods and applications, and even new journals and commercial service providers. Despite widespread recognition of its great promise to aid decision-making in environmental management, the applied use of metabarcoding still requires improvements to reduce the multiple errors that arise during PCR amplification, sequencing, and library generation.
Biodiversity soup II demonstrates a co-designed wet-lab and bioinformatic workflow for the metabarcoding of bulk samples. The authors show how to (i) eliminate sample misassignment due to tag-jumping, (ii) reduce false-negatives and taxonomic bias (‘drop-outs’), and (iii) reduce false-positive detections (‘drop-ins’). Furthermore, the authors find no evidence of tag bias during PCR, which will allow researchers to continue using tagged primers and thus reduce contamination risk.
To aid learning, reproducibility, and the design and testing of alternative metabarcoding pipelines, the authors provide the Illumina and input species reference sequence datasets, accompanying scripts, and a spreadsheet to aid primer tag design. This paper will help the community apply metabarcoding more reliably.
This study was financially supported by grants from the State Key Laboratory of Genetic Resources and Evolution and Center for Excellence in Animal Evolution and Genetics, Strategic Priority Research Program of the Chinese Academy of Sciences and National Natural Science Foundation of China, etc.
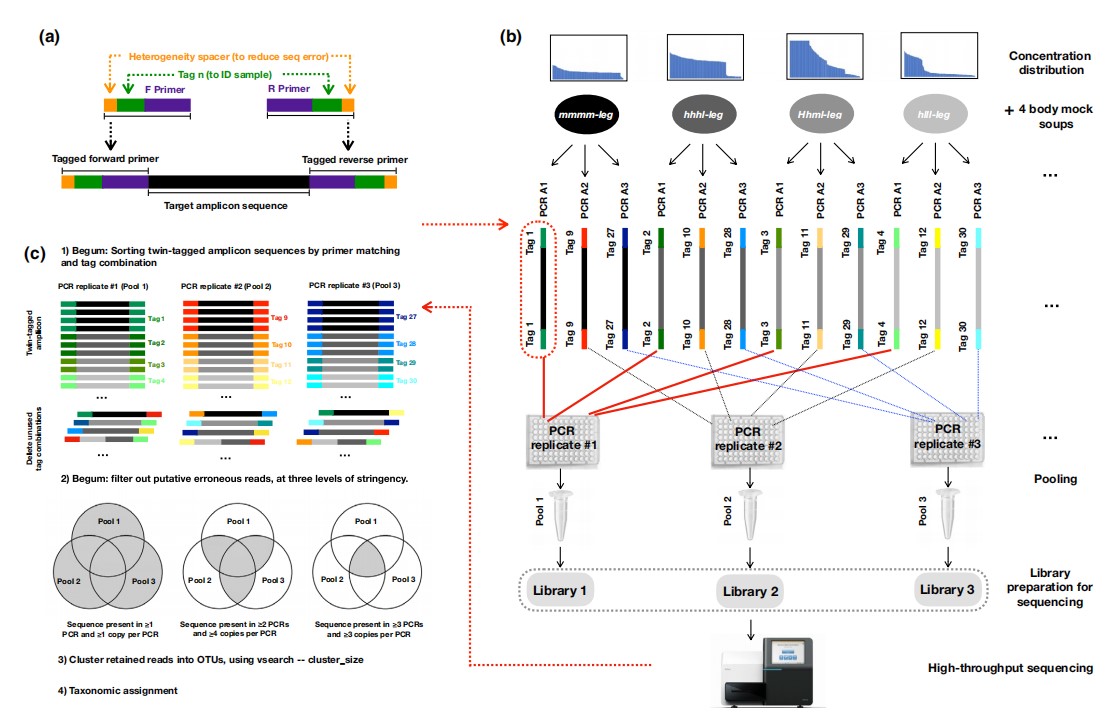
Schematic of study. (a) Twin-tagged primers with heterogeneity spacers (above) and final amplicon structure (below). (b) Each mock soup (e.g. Hhml-leg) was PCR amplified three times (1, 2, 3) under a given PCR condition (A–H). Each of the three PCRs per soup used a different twin tag, following the Begum strategy. There were eight mock soups (Hhml/hhhl/hlll/mmmm X body/leg), where H, h, m and l indicate different DNA concentrations (details in Figure 2). PCR replicates 1 from each of the eight mock soups were pooled into the first amplicon pool (solid red lines), PCR replicates 2 were pooled into the second amplicon pool (black dashes) and PCR replicates 3 were pooled into the third amplicon pool (blue dashes). The entire set-up in B was repeated eight times for the eight PCR experiments (A–H), which thus generated (3 × 8 =) 24 sequencing libraries. (c) Key steps of the Begum bioinformatic pipeline. For clarity, primers and heterogeneity spacers not shown.
(By YANG Chunyan, Editor YANG Yingrun)
Contact:
YANG Chunyan
yangyahan@mail.kiz.ac.cn
